物理学家热衷于在软件中重塑世界。模拟可以让你探索现实的多种版本,以寻找模式或测试可能性。但如果你想要一个精确到单个原子和电子的模拟,你很快就会耗尽计算资源。
机器学习模型可以近似详细的模拟,但通常需要大量昂贵的训练数据。一种新方法表明,物理学家可以将他们的专业知识应用于机器学习算法,帮助它们在少量包含几个原子的模拟上进行训练,然后预测包含数百个原子的系统的行为。未来,类似的技术甚至可以表征包含数十亿个原子的微芯片,在故障发生之前预测它们。
研究人员从模拟的 16 个硅和锗原子单元开始,这两种元素通常用于制造微芯片。他们使用高性能计算机来计算原子电子之间的量子力学相互作用。给定原子的特定排列,模拟会生成单元级特征,例如它的能带,即其电子可用的能级。但“你意识到,在我们能够使用第一性原理方法研究的玩具模型和现实结构之间存在着巨大的差距,”科罗拉多大学博尔德分校的物理学家、论文的资深作者桑加米特拉·内奥吉说。她和她的合著者阿特姆·皮马切夫能否利用机器学习弥合这一差距?
这项发表在 6 月份的《npj 计算材料》上的研究,其想法是训练机器学习模型从 16 个原子的排列中预测能带,然后将这些模型输入更大的排列,看看它们是否可以预测它们的能带。“本质上,我们试图探究这个包含数十亿个原子的世界,”内奥吉说。“那里的物理学是完全未知的。”
内奥吉说,传统的模型可能需要一万个训练样本。但她和皮马切夫认为他们可以做得更好。因此,他们应用物理原理来生成正确的训练数据。
首先,他们知道应变会改变能带,因此他们模拟了具有不同应变量的 16 个原子单元,而不是浪费时间生成大量具有相同应变的模拟。
其次,他们花了一年时间寻找一种描述原子排列的方法,这种方法对模型有用,一种“指纹”单元的方法。他们决定将一个单元表示为一组具有平坦壁的 3D 形状,每个原子一个。形状的壁由原子与其邻居之间等距的点定义。(这些形状紧密地组合在一起,形成所谓的 Voronoi 镶嵌。)“如果你足够聪明,可以创建一组好的指纹,”内奥吉说,“那就消除了对大量数据的需求。”他们的训练集最多包含 357 个样本。
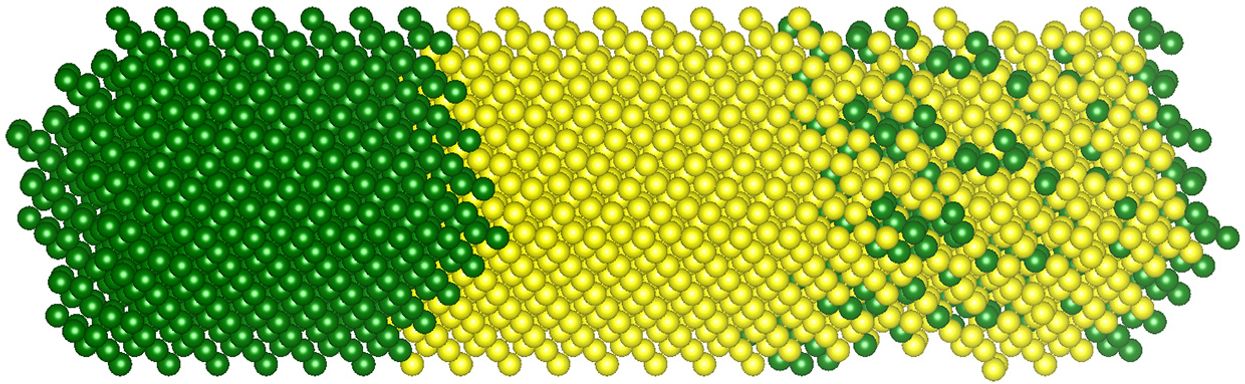
内奥吉和皮马切夫训练了两种不同类型的模型——神经网络和随机森林,或一组决策树——并在三种不同类型的结构上测试了它们,将它们的数据与详细模拟的数据进行比较。第一个结构是“理想超晶格”,它可能包含几层纯硅原子层,然后是几层纯锗原子层,依此类推。他们在应变和松弛条件下测试了这些结构。第二个结构是“非理想异质结构”,其中给定层在厚度上可能会有所不同,或者包含缺陷。第三个是“制造的异质结构”,它具有纯硅部分和硅锗合金部分。测试用例包含多达 544 个原子。
在各种条件下,随机森林的预测与模拟输出的差异为 3.7% 到 19%,神经网络的差异为 2.3% 到 9.6%。
“我们没有预料到能够模拟如此大的系统,”内奥吉说。“五百个原子是一个巨大的数字。”此外,即使系统中原子数量呈指数级增长,模型进行预测所需的计算时间也仅呈线性增长,这意味着包含数十亿个原子的世界是相对可达的。
“我认为这非常聪明,”伊利诺伊州莱蒙特阿贡国家实验室的计算科学家洛根·沃德谈到这项研究时说。“作者在不同阶段巧妙地将他们对物理学的理解结合起来,使机器学习模型能够工作。我以前从未见过类似的东西。”
在内奥吉即将发表的后续工作中,她的实验室进行了逆向操作。给定材料的能带,他们的系统预测了它的原子排列。这样的系统使他们更接近于诊断计算机芯片中的故障。如果半导体的电导率出现问题,他们可能会指出缺陷所在。
他们提出的框架也适用于其他类型的材料。关于生成和表示训练样本的物理信息方法,内奥吉说,“如果我们知道在哪里寻找,一点点信息就能告诉我们很多东西。”